El retinoblastoma es de las enfermedades más comunes en la infancia, este
se desarrolla como un tumor intraocular maligno. Sugiere que se origina de células
retinianas primitivas capaz de diferenciarse de células gliales como
neuronales.
El gen
más relacionado con esta patología es el gen RB1, este forma parte de la
familia de los genes supresores de neoplasias, los cuales inhiben el
crecimiento celular y regulan negativamente la proliferación. El gen RB1 tiene una
longitud de 180,388 pares de bases y está formado por 27 exones, que codifican
para un ARNm que se traduce en un fosfoproteína nuclear (PRB),constituida por
928 aminoácidos, con un peso de 105 -110 kDa. PRB es una proteína reguladora
del ciclo celular, en donde su forma hipofosforilada suprime la transcripción
de genes que regula la división celular, por lo que está involucrada en el
ciclo celular y en el proceso de apoptosis. El gen RB1 se localiza en el
cromosoma 13 en la región q 14.2.
En los
casos en que el gen RB1 se encuentra normal, es probable que otros factores o
proteínas (factores de transcripción como la familia E2F desacetilasas de
histonas, desaminasas, cinasas, ciclinas etc) que regulan la función de
proteína pRB, sean los que estén alterados favoreciendo de manera indirecta el
desarrollo de retinoblastoma. Se han reportado todo tipo de mutaciones en el
gen RB1 (eliminaciones, inserciones, etc) siendo las más frecuentes las
mutaciones puntuales, estas se presentan en un 50% de las alteraciones de este
gen. La mayoría de los estudios concuerdan con que los exones 3, 8, 18, 19 y 20
son las regiones preferenciales de mutación, también llamadas hot spots. La
mayoría de las mutaciones son transiciones de C a T (De citosina a timina),
aunque no se ha comprobado, se propone que estas citosina están hipermetiladas,
permitiendo su desaminación generando así mutaciones, por lo que se cree que
este pudiera ser uno de los principales mecanismos que generan mutaciones en
RB1. Esta mutación generará una ganancia de Treonina, el cual es un aminoácido
susceptible a ser fosforilado.
Se ha manifestado
que algunas oncoproteinas virales como el antígeno E1A tipo 5 del adenovirus
humanos y E7 del VPH de alto riesgo, forman complejos con pRB, esta proteína
además de ser secuestrada es marcada para su degradación por el sistema de
ubiquitina. Dicha interacción provoca alteraciones en el control del ciclo
celular permitiendo que la célula permanezca dividiéndose descontroladamente.
Tumores intraoculares. Retinoblastoma. Dr. García-Arumí de IMO Barcelona.
Bibliografia
AbramsonDH, BeaversonK, SanganiPy cols. La
detección deretinoblastoma: la
presentación designoscomopronosticadoresdepacientey la supervivenciaocular.Pediatrics2003;112(6): 1248.
Las más de 30 000
millones de células que constituyen nuestro organismo nacen, crecen, se dividen
y mueren bajo la estricta vigilancia del material hereditario, o sea, de la
molécula de ADN. Por tanto unas células regulan la proliferación de otras, para
asegurarse de este modo que los órganos y tejidos crezcan en equilibrio y
mantengan la arquitectura corporal. La reproducción celular está supervisada
por determinados sistemas de control extremadamente rigurosos. Para que una
célula se divida en 2 células hijas idénticas es necesario la participación de
una gran cantidad de moléculas como proteínas, enzimas, factores de
crecimientos y de genes que se activan y desactivan con la precisión de la
maquinaria de un reloj. En las células normales, el reloj integra la mezcla de
señales reguladoras del crecimiento recibidas por la célula y decide si ésta
debe o no pasar a través de su ciclo de vida. El más mínimo fallo que tenga
lugar en uno de los sistemas de control puede acarrear una tragedia celular.
Las células de un tumor
descienden de una ancestral común, que en algún momento, generalmente décadas
antes de que el tumor se manifieste, inició un programa de división indebido.
La transformación maligna de una célula acontece por acumulación de mutaciones
en unos genes específicos, los cuales son la clave molecular para entender las
raíces del cáncer.
Estos genes están
agrupados en 2 familias. La primera está integrada por los protooncogenes, los
cuales dirigen la producción de proteínas como ciclinas, factores de
crecimiento, receptores, etcétera que estimulan la proliferación celular.
Cuando éstos mutan se transforman en oncogenes, los cuales son capaces de
orquestar la multiplicación anárquica de las células, de modo que algunos de
ellos hasta fuerzan la maquinaria celular plara que sintetice de forma masiva
determinados factores de crecimiento.
La segunda familia está
integrada por los genes supresores de tumores también conocidos como genes
supresores, que en el organismo sano controlan la proliferación celular. Ellos,
por tanto son reguladores negativos de crecimiento y cuando no están presentes
en la célula o se encuentran inactivos a causa de mutaciones, las células dejan
de crecer normalmente y adquieren propiedades proliferativas anormales,
características de las células tumorales.
Algunos genes supresores y su asociación con los diferentes
tumores
A diferencia de aquellos
tumores causados como resultados de alteraciones de los oncogenes, donde una mutación
que active un simple alelo es dominante sobre su variante sana y la
tumorigénesis resulta de la ganancia de una función, existen tumores que son
causados por un mecanismo diferente como la pérdida de ambos alelos en un locus
(lo cual tiene acción tumorigénica). La propensión para formar tales tumores
puede ser heredado a través de la línea germinal y esto también puede ocurrir
como resultado de cambios somáticos en el individuo. Tales casos identifican
genes supresores de tumores: secuencias genómicas cuyos productos son
necesarios para el funcionamiento normal de la célula y cuya pérdida de función
causa tumores.
En el conocimiento de
los genes supresores se han dado algunos pasos importantes. Los estudios
moleculares han identificado hasta la fecha más de 17 genes supresores de
tumores implicados directamente en el cáncer humano. Ellos codifican para una
serie de proteínas localizadas en distintas regiones dentro de la célula, tanto
en el citoplasma como en el núcleo.
Los 2
genes mejor caracterizados de esta clase codifican para las proteínas p53 y RB.
Genes
supresores de tumores
Genes para
proteínas en el citoplasma
APC Está
involucrado en cánceres de colon y estómago.
DPC4
Codifica para una molécula en una ruta de señalización que inhibe la división
celular. Involucrado en cáncer pancreático.
NF-1
Codifica para una proteína que inhibe una proteína (Ras) estimulatoria.
Involucrado en neurofibroma y pheochromocytoma (cánceres de el sistema nervioso
periférico) y leucemia mieloide.
NF-2
Involucrado en meningioma y ependimoma (cánceres de cerebro) y schwannoma
(afecta la vaina que envuelve los nervios periféricos).
Genes para
proteínas en el núcleo
MTS1
Codifica para la proteína p16, un componente del reloj del ciclo celular.
Involucrada en un amplio rango de cánceres.
RB
Codifica para la proteína pRB, uno de los principales controles del ciclo
celular. Involucrado en el retinoblastoma y cánceres de hueso, vejiga, células
pequeñas de pulmón y cáncer de mama.
p53
Codifica para la proteína p53, la cual puede detener la división celular e
inducir a las células anormales a matarse ellas mismas. Involucrado en una gran
cantidad de cánceres.
WT1
Involucrado en el tumor de Wilm del riñón.
Genes para
proteínas cuya localización celular no está clara aún
BRCA1
Involucrado en cánceres de mama y ovario.
BRCA2
Involucrado en cáncer de mama.
VHL
Involucrado en cáncer de células renales.
p53
El gen p53 es
considerado por muchos autores como "el guardián del genoma". A
partir de este gen se sintetiza una proteína, que lleva el mismo nombre y se
activa cuando la célula se dispone a dividirse, para vigilar la secuencia
normal de acontecimientos genéticos que permiten la proliferación celular. Si
el material genético de la célula resulta dañado o si algún sistema de control
se desajusta, esta lo detecta e intenta restaurarlo. Si la lesión no es grave,
la p53 detiene la división celular y activa los genes reparadores del ADN. Si
la p53 estima que el daño es irreparable entonces ordena que se pongan en
marcha los mecanismos genéticos para que la célula entre en apoptosis o muerte
celular programada. Si este gen (p53) sufre alguna mutación, no permite que la
célula sea eliminada mediante la muerte programada, tampoco se ocupa de reparar
los daños en el ADN y da lugar al inicio del proceso tumoral. Este gen es el
más frecuentemente mutado en los cánceres humanos, más de un 50 % de los
tumores tienen genes p53 anormales, produciéndose una proteína alterada.
Pero la pérdida de la
función de esta proteína no solo puede deberse a una mutación en el gen que la
origina, sino que existen otros mecanismos que pueden provocar que la célula
carezca de un control tan importante como éste. Un ejemplo bien estudiado es la
infección por ciertos virus como el papilomavirus humano, el cual presenta una
proteína temprana denominada E6, la cual se une a la proteína p53 y potencia su
degradación mediada por ubiquitina.
RB
El producto del gen
supresor de tumores RB, ejerce su efecto durante la primera parte de la fase G1
del ciclo celular. En este período o en las células quiescentes, esta proteína
es unida al factor de la transcripción E2F. Este complejo tiene 2 funciones, en
primer lugar, muchos de los genes cuyos productos son esenciales para la fase S
de dicho ciclo dependen de la actividad del factor E2F. Por tanto el RB,
mediante el secuestro de este factor de la transcripción garantiza que la fase
S no pueda ser iniciada. En segundo lugar, el complejo E2F-RB reprime la
transcripción de otros genes. En el punto de restricción de la fase G1 del
ciclo celular o cerca del mismo el RB es fosforilado por el complejo quinasa/
dependiente de ciclinas y esta fosforilación causa la liberación del factor E2F
por el RB, el cual entonces activa los genes cuyas funciones son requeridas
para la fase S, también derreprime otros genes cuya función estaba controlada
por el mismo complejo.
El retinoblastoma es una
enfermedad humana infantil que involucra un tumor de retina. La misma es
causada por la pérdida de ambas copias del gen RB en la banda q14 del cromosoma
133,18,19 En la forma hereditaria un cromosoma tiene una deleción en esta
región y la segunda copia es perdida por deleción somática en el individuo. En
la forma esporádica, ambas copias se pierden por eventos somáticos
individuales. (fig.). Las deleciones en los alelos normales del gen RB no es la
única causa de la pérdida de la función proteica. También los papilomavirus
humanos se valen de una proteína temprana, denominada E7, capaz de unir a la
proteína RB permitiendo la liberación del factor de la transcripción E2F con la
activación de los genes cuyos productos proteicos son requeridos en los
procesos de síntesis celulares que acontecen mientras la célula se prepara para
su división.
NF1
Otra de las formas en
que pueden actuar estos genes supresores de tumores es bloqueando el flujo de
señales a través de los circuitos estimulatorios del crecimiento. Uno de estos
genes supresores de tumores es el producto proteico del gen NF1. Esta proteína
citoplasmática atrapa a la proteína Ras antes de que esta pueda emitir sus
directivas promotoras del crecimiento. Las células carentes de NF1, han perdido
un contrabalance importante para Ras. Este gen está relacionado con los
neurofibromas, pheochromocytoma, leucemia mieloide y ciertos cánceres del
sistema nervioso periférico.
El retinoblastoma es
causado por la pérdida de ambas copias del gen RB en la banda del cromosoma
13q14. En la forma hereditaria, un cromosoma tiene una deleción en esta región,
y la segunda copia es perdida por mutación somática en el individuo. En la
forma esporádica ambas copias se pierden por eventos somáticos individuales.
BRCA1
En el año 1990, un grupo
de investigadores reportó la relación existente entre la aparición temprana del
cáncer de mama con una región del brazo largo del cromosoma 17. Más tarde se
precisó que la región que contenía el locus de la enfermedad (denominado BRCA1)
era en el 17q21. En 1994 se reportó la clonación y la secuenciación del gen
BRCA1. Se conoce de la existencia de cientos de mutaciones diferentes de las
cuales pueden resultar proteínas truncadas o ausentes. Se han descrito también
5 mutaciones puntuales en tumores de ovario. Además, se ha detectado la pérdida
de heterocigosidad de 2 nuevos genes supresores de tumores.
BRCA2
En el brazo 13q fue
mapeado otro locus relacionado con la aparición del cáncer mamario familiar. A
finales de 1995 fue identificado el gen y la secuencia completa fue publicada en
marzo del siguiente año. Mutaciones en BRCA2 están muy relacionadas con el
cáncer de mama, siendo las mutaciones somáticas de este gen infrecuentes en la
aparición del cáncer de ovario.
Detección de alteraciones moleculares en los genes supresores
de tumores
En la actualidad se
cuenta con técnicas muy útiles en la determinación de alteraciones moleculares
en los genes supresores de tumores. La pesquisa de la pérdida de
heterocigosidad permite la detección de inserciones o deleciones. Si se trata
de cambios pequeños en la secuencia nucleotídica como mutaciones puntuales y
pequeñas deleciones e inserciones que no afecten la transcripción y la
traducción de la proteína resultan de gran utilidad el análisis de polimorfismo
conformacional de simple cadena (SSCP, del inglés single-stranded conformation
polymorphism), la electroforesis en geles con gradientes desnaturalizantes
(DGGE, del inglés denaturing gradient gel electrophoresis) y los ensayos de
truncamiento de la proteína (PTT, del inglés protein truncation). Una vez que
se ha visto la presencia de mutaciones en las muestras, entonces la
secuenciación directa determinaría la naturaleza de la mutación.
Importancia de los genes supresores de tumores en el
diagnóstico y en la terapia
Uno de los factores
limitantes en los tratamientos utilizados para la cura del cáncer es la
toxicidad o daño que se le hace a los tejidos normales. Las altas dosis de
radiaciones y agentes quimioterapéuticos necesarias para matar células
tumorales resistentes podría conducir a la muerte del paciente como resultado
de la toxicidad sobre los tejidos normales. El éxito consiste en encontrar la
forma de matar selectivamente las células tumorales sin afectar al tejido
normal. Para esto es muy importante conocer las diferencias moleculares y
celulares entre células normales y células tumorales con vista a definir
blancos específicos dentro de estas últimas. La terapia génica abre las puertas
a una nueva era, aunque los estudios en humanos apenas comienzan, realizándose la
mayoría de dichos experimentos en animales, donde se han obtenido resultados
alentadores. Uno de los principales objetivos de la transferencia de genes
terapéuticos contra el cáncer es normalizar el ciclo celular inhibiendo
oncogenes o restaurando la actividad de los genes supresores de tumores. En
personas que hayan perdido ambos alelos del gen que codifica para la proteína
p53 o para RB, la administración de las versiones sanas pueden restablecer el
funcionamiento normal de la célula, la cual contaría otra vez con su sistema de
vigilancia de los eventos genéticos que tienen lugar durante la proliferación
celular.32 Por esta razón los estudiosos del tema se enfrascan en la difícil
tarea de determinar y caracterizar todas las variantes genéticas implicadas en
la aparición y desarrollo del cáncer.
BIBLIOGRAFIA
Rev
Cubana Oncol 2001;17(1):65-71
Instituto Nacional de Oncología y Radiobiología
Hortwell LH, Kastan MB. Cell cycle control and
cancer. Science 1994;266:1821.
Kastan MB. Molecular biology of cancer: the cell
cycle. En : de Vita VT. Cancer: principles and practice of oncology. 5 ed.
Philadelphia: Lippincot , 1997;121.
Lewin B. Oncogenes and cancer. En: Lewin B. Genes VI. 6 ed. New York: Oxford University,
1997;1131-72.
Alberts B, Bray D, Lewis J, Raff M, Roberts K,
Watson JD. Molecular biology of cell. 3 ed. New York: Garland 1994:1255.
Boss JL, Kreijl CF van. Gene and gene products
that regulate proliferation and differenciation: critical targets in
carcinogenesis. IARC Sci Publ 1992;116:57-67
MODOS DE ACCIÓN DE LOS ONCOGENES EN TUMORES HUMANOS
ASOCIADOS
A. FACTORES DE CRECIMIENTO
Las células normales requieren de la estimulación de
factores de crecimiento para proliferar. Muchas células cancerosas, adquieren
la capacidad para sintetizar los mismos factores de crecimiento a los que
responden, generando un ciclo autócrino. Por ejemplo muchos glioblastomas
secretan factores de crecimiento derivados de las plaquetas (PDGF) y expresan
el receptor PDGF y muchos sarcomas forman tanto el factor de crecimiento
transformante α (TGF-α) como su receptor.
La producción incrementada de los factores de crecimiento
no es suficiente para la transformación neoplásica, pero la proliferación
conducida por factores de crecimiento contribuye al fenotipo maligno mediante
un incremento del riesgo de mutaciones espontáneas o inducidas en la población
celular en proliferación
Con respecto al modo de acción de los factores de
crecimiento podemos decir que el ligado a sus receptores de membrana en las
células blanco, activa señales de quinasa que conducen a la activación del
ciclo celular y por tanto a proliferación.
Como ejemplo mencionamos al Factor de Crecimiento derivado de plaquetas (PDGF)
es un producto de oncogén que actúa normalmente de este modo.
Modelo de la acción de los genes RAS. Cuando una célula normal es
estimulada a través de un receptor de crecimiento, RAS inactivo unido a GDP, se
activa hasta un estado unido a GTP. Ras activado recluta RAF y estimula la vía
de la MAP-cinasa para transmitir señales promotoras del crecimiento al núcleo.
La proteína RAS mutada está permanentemente activada debido a la incapacidad
para hidrolizar la GTP, conduciendo a una estimulación continua de las células
sin ningún desencadenante externo. El anclaje de RAS a la membrana celular
mediante la molecula de farsenil es esencial para su acción.
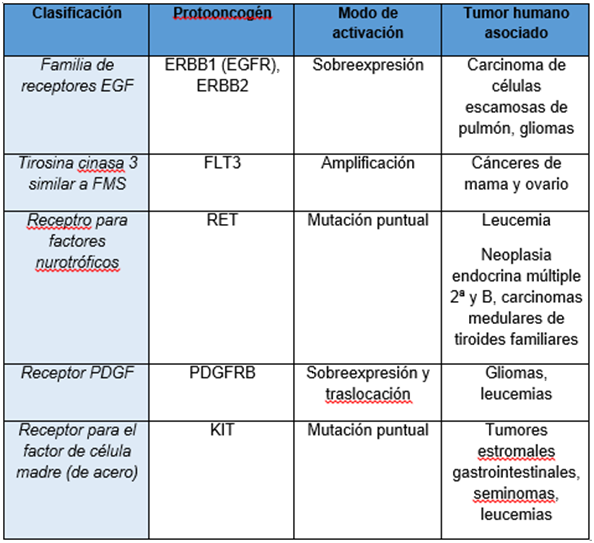
B. RECEPTORES DE FACTORES DE CRECIMIENTO
Los receptores de factores
de crecimiento, que ocupan la posición siguiente en la cascada de señales que
va desde el exterior celular al interior para la regulación de su crecimiento,
diferenciación y apoptosis, pueden ser otro de los eslabones que fallan en un
proceso canceroso. Un ejemplo importante dentro del grupo de protooncogenes que
codifican receptores de factores de crecimiento es el gen erbB, que se encuentra frecuentemente amplificado en
tumores humanos. El oncogén erbB deriva
del gen celular normal, que codifica el receptor de membrana del factor de
crecimiento epidérmico (EGF). Su activación por amplificación génica se da en
un amplio número de tumores, generalmente provoca una sobreexpresión de su
producto, por lo que se producen elevados niveles del receptor.
El oncogén erbB se puede activar también por la deleción del
dominio de unión de su ligando, en el extremo amino terminal, como erbB-2, kit, met, ret y trk. Así pues, la versión oncogénica del erbB produce un receptor sin la región de unión a
su ligando (el EGF). Aunque el erbB
oncogénico presenta también otras mutaciones, parece ser que es esta deleción
amino-terminal la principal responsable de su capacidad para transformar
células normales en cancerosas. En la ruta normal, la unión del EGF regula la
actividad tirosina quinasa del receptor, promoviendo las acciones posteriores
de la cascada, de forma que en ausencia del EGF no hay actividad tirosina
quinasa. En su versión oncogénica se produce una activación constitutiva de
esta actividad tirosina quinasa de proteínas, independientemente de que se una
el EGF o no. Por tanto, esta continua actividad fosforilando proteínas de la
cascada sería la responsable de la proliferación anormal que conduciría a una
neoplasia.
Se
han encontrado varios oncogenes que codifican receptores de F.C. Las mutaciones
pueden ser de dos tipos:
que se trunque el
receptor.
mutaciones puntuales.
Los
receptores más afectados son para EGF.
Puede ocurrir que una
alteración en el ADN desencadene la síntesis de una proteína anormal cuya
conformación se vuelva constitutivamente activa y no responda a ningún tipo de
señal o control externo. Se conocen otros casos en que la activación del oncogén
provoca la dimerización y consiguiente activación permanente de las subunidades
de receptores de membrana y sus cascadas de señales estimuladoras del
crecimiento.
C. PROTEÍNAS
IMPLICADAS EN LA TRANSDUCCIÓN DE SEÑAL
Existen varias proteínas que tienen como
finalidad convertir las señales externas que se presentan. Para esto están
constituidas por varios dominios, actividades de enlaces y actúan por medio de
moléculas se señalización intracelular.
Cuando un ligando
se une a un receptor provoca una serie de acontecimientos mediantes los cuales
las señales provenientes de exterior de la célula son traducidas por estas
proteínas al interior. Estos receptores pueden ubicarse ya sea en la superficie
de la célula diana como también en el citoplasma o incluso en el núcleo.
Entre los receptores y mecanismos de transducción de señal
encontramos:
Receptores con actividad tirosina
cinasa intrínseca:Los receptores tiene un dominio de unión al ligando
extracelular, una región transmebrana y una cola citoplasmática dotada de
actividad tirosina cinasa intrínseca. La unión del ligando determina la
dimerización del receptor, la fosforilación de la tirosina y la activación del
receptor tirosina cinasa.
Receptores sin actividad tirosina
cinasa intrínseca que reclutan cinasas: Los ligandos para estos receptores incluyen muchas
citocinas, como IL-2 e IL-3, y otras interleucinas. Estos receptores transmiten
señales extracelulares hacia el núcleo mediante la activación de los miembros
de la familia de proteínasJAK(cinasa Janus).
Receptores acoplados a proteína G: transmiten señales a la célula mediante
proteínas triméricas que se unen al GTP y supone la familia más extensa de
receptores de la membrana plasmática. La unión del ligando induce cambios en la
forma de los receptores y determina su actividad.
Receptores de las hormonas esteroideas: suelen localizarse en el núcleo y se comportan como
factores de transcripción dependientes de ligandos.
Las oncoproteínas
que imitan la función de las proteínas traductoras de señales citoplasmáticas
descritas anteriormente están situadas en su mayoría en la capa interna de la
membrana plasmática, donde reciben señales del exterior de la células.
Bioquímicamente son heterogéneas. El ejemplo mejor estudiado de una
oncoproteína transductora de señal es la familia RAS de proteínas que se unen
al GTP.
Las proteínas RAS oscilan
entre un estadio excitado de transmisión de la señal y un estadio quiescente,
donde el inactivo se encuentra al estar unido al GDP. El ciclo ordenado de la
proteína RAS depende de dos reacciones: 1) intercambio de nucleótido (GDP por
GTP), que activa la proteína y 2) hidrólisis del GTP, que lo inactiva. El RAS
activado estimula los reguladores de la proliferación cinasa activada por
mitógenos (MAP) que inundan el núcleo con señales de proliferación.
Existen tres
tipos de RAS: (HRAS, KRAS, NRAS)
Alteraciones de
las tirosinas cinasas sin receptor: Se puede producir una actividad oncógena
debido a mutaciones en la actividad de este receptor, los cuales normalmente
intervienen en las vías de transducción de la señal que regulan el crecimiento
celular.
D. PROTEÍNAS REGULADORAS NUCLEARES
El gen c-myc es
uno de los más importantes proto-oncogenes que codifican proteínas nucleares.
Los productos proteicos del c-myc y otros genes myc,
así como de muchos otros protooncogenes, regulan la transcripción de una
cohorte de genes diana en el núcleo uniéndose a la secuencia de ADN de estos
últimos, permitiendo o impidiendo su transcripción.
La familia de genes Myc contiene
al menos siete genes relacionados de forma estrecha,Myc, Nmyc, Lmyc, Pmyc, Rmyc, Smyc y Bmyc,
estos genes codifican factores de transcripción del tipo cremallera de leucina,
de forma que están relacionados con la expresión de determinados genes. La
proteína myc está presente en una amplia variedad de tejidos
adultos y en todos los estadios durante el desarrollo embrionario.
El gen myc está
implicado en el control de la proliferación normal, transformación y
diferenciación; el myc en células no transformadas es un
factor de crecimiento esencial para la progresión dentro del ciclo celular.
Niveles elevados de su producto génico aceleran el crecimiento, mientras que
una downregulation de la expresión del myc normalmente se
corresponde con el comienzo de la diferenciación y su expresión constitutiva
interfiere con la diferenciación normal.
Otro ejemplo de proteínas
nucleares relacionadas con el control del reloj celular es la codificada por el
gen erbA, que es un receptor de hormonas tiroideas. Es interesante
destacar que el erbA parece no inducir directamente la
transformación maligna por sí mismo, sino que potenciaría la capacidad
transformante del erbB en células eritroides.
La mayor diferencia entre
la proteína del oncogén erbA respecto al receptor de hormonas
tiroideas normal es que ha perdido la posibilidad de unir hormonas tiroideas,
presumiblemente como consecuencia de múltiples mutaciones en su dominio
carboxi-terminal de unión a su ligando. Así, la proteína de la versión oncogénica
del erbA actúa como un represor constitutivo de genes
inducibles por hormonas tiroideas independiente del control de tales hormonas.
El gen Bcl-1 es
un oncogén que codifica la ciclina D1. En varias transformaciones malignas de
las células B, en leucemia linfoide crónica y la leucemia mieloide múltiple, se
produce la activación del oncogén ras por translocación entre
los cromosomas 11 y 14. En algunas líneas celulares de cáncer de mama se ha
podido observar la sobreexpresión de este gen o un aumento de la estabilidad
relativa de los transcriptos del mismo. La ciclina D1 activa la cdk-4, que
interviene en la fosforilación del pRB y da como resultado un aumento de la
proliferación celular.
Así mismo también se hace
mención de las proteínas histonas, que
son proteínas reguladoras de genes nucleares. Encargadas del empaquetamiento
del ADN nuclear, estas proteínas están capacitadas para ejercer una doble
misión: bloquear la activación de muchos genes o promover, por contra, su
expresión cuando existe una mutación o peligro de formación de una neoplasia.
Las histonas son unas proteínas
pequeñas que están en el núcleo. Som muy básicas lo que les facilita unirse al
ADN para ejercer su función de empaquetarlo formando parte de la cromatina.
Las histonas son de dos tipos: H1 (ó H5) y las
histonas nucleosómicas. Las histonas nucleosómicas son más pequeñas ( 102 a 135
aminoácidos) y forman los nucleosomas al enrollar ADN sobre un grupo de ellas.
Las histonas nucleosómicas son la H2A, H2B, H3 y H4 y están muy conservadas a
lo largo de las diferentes especies.eucariotas. En el núcleo de la célula hay
un gran número de ellas (alrededor de 60 millones de cada tipo). Las histonas
pueden ser modificadas tras la traducción lo que les cambia sus propiedades de
unión al ADN y a proteínas nucleares.
Las histonas H3 y H4 tienen largas colas N-terminales hacia el
exterior del nucleosoma que son susceptibles de ser modificadas covalentemente.
Las modificaciones que pueden sufrir las histonas son: acetilación, metilación,
fosforilación y mono-ubicuitinación y sumoilación. Estas modificaciones pueden
ser heredadas, influyen en la expresión génica, cambian la arquitectura local
de la cromatina y podrían también reclutar otras proteínas que reconozcan
modificaciones específicas de las histonas según la hipótesis llamada el
`código de las histonas. Existe una correlación entre la acetilación de
histonas y aumento de transcripción que parece debido a que tras acetilarse la
histona se une menos al ADN.
Por otra parte parece haber activadores de la transcripción que se unen
específicamente a acetil-lisina mediante un módulo llamado `Bromo`. La
metilación de histonas puede tanto activar como reprimir la transcripción
dependiendo de qué residuos de lisina en qué histonas son metilados. Estos
residuos de metil-lisina parece que reclutan proteínas que contienen un dominio
de unión a ellas llamado `Cromo`.
La acetilación de las histonas regula la expresión de genes
relacionados con la inflamación y también parece tener un papel en diversas
funciones tales como reparación de ADN y proliferación celular, por lo que se
plantea usar inhibidores de la histona deacetilasa como nuevos agentes
antiinflamatorios. La deacetilasa de histonas actúa como represor de la
transcripción a través de interacciones con otras proteínas lo que lleva a
remodelación de la cromatina. Hoy día parece claro que determinados patrones de
modificación de histones conducen a determinadas patologías entre las que
podrían encontrarse, además de patologías tumorales, patologías inflamatorias
de localización broncopulmonar como el asma.
E. REGULADORES DEL CICLO CELULAR
Control del ciclo
celular
El funcionamiento correcto de los procesos del ciclo
celular requiere de cambios en complejos enzimáticos, entre los que se
encuentran las ciclinas, las cinasas dependientes de ciclinas (CDK) y los
complejos que se forman ent re ambas (CDK-ciclina). Las formas activas de los
complejos CDK-ciclina están constituidas de dos proteínas (una cinasa y una
ciclina). Las cinasas son enzimas que realizan la fosforilación de proteínas, y
este evento es de gra n importancia para la regulación del ciclo celular. Los complejos
CDK-ciclina dirigen a la célula de una fase a otra del ciclo celular. Por lo
tanto, la dinámica del ciclo dependerá de las formas activas o inactivas de los
complejos CDK-ciclina, entre otros muchos sucesos. Aunque inicialmente se
estudió la función de los complejos CDK-ciclina en Saccharomyces cerevisiae y
Saccharomyces pombe, se han identificado enzimas con actividades similares en
mamíferos. Los estudios en levaduras aún emplean los términos de p34cdc2 para
CDK1; sin embargo, las funciones en el ciclo celular son idénticas. Se sabe que
CDK4, CDK2 y CDK5 se expresan conjuntamente con las ciclinas D1, D2, D3, E, A y
B, durante la progresión de la fase G1 a la fase M (mitosis). El complejo
CDK4-D funciona tempranamente en respuesta a factores de crecimiento. Los
complejos CDK2-E y CDK2-A son esenciales para la replicación del ADN, y los
comple jos CDK2-A y CDK2-B son importantes para la mitosis. Recientemente se
han reportado CDK adicionales. La mayoría de los complejos CDK-ciclina de
mamíferos pueden remplazar funcionalmente los correspondientes complejos de
levadura, y lo mismo ocurre para las enzimas que regulan la actividad de las
cinasas, lo que sugiere que por la importante función que tienen los complejos
CDK-ciclina en el ciclo celular, éstos se han conservado durante la evolución
de los eucariontes.
Cuando existe algún daño genético, los mecanismos de
control transcripcional de los complejos CDK-ciclina inducen la interrupción
del ciclo celular hasta que el daño se corrige. Esto ocurre en S. ce revisiae y
en oocitos de Xenopus. En mamíferos, la interrupción de la proliferación de la
línea celular Mu1Lu por TGF-b1 es mediado por la proteína p27 que evita el
ensamblaje y activación del complejo CDK2-E.7 Además, se han identificado en
mamíferos otras dos proteínas inhibidoras de la actividad de los complejos
CDK-ciclina: p16 y p21; 8-11 las cuales bloquean la progresión del ciclo
celular en la fase G1; aunque pueden tener otras funciones aún no conocidas. La
inducción de p21 (WAF1 o CIP1) depende de p53 y ocurre cuando las células
tienen daño en el ADN. La proteína p21 interfiere con la actividad de cinasa
del complejo CDK-E. La proteína p16 inhibe la activación de CDK4-D1. Durante la
transición de la fase G1 a la fase S, diferentes substratos pueden ser blancos
de los complejos CDK2-E, como por ejemplo la proteína supresora de tumores pRb.
La proteína pRb se asocia con E2F durante la fase G1 y cuando pRb se fosforila
libera a E2F, el cual participa en la transcripción de varios genes requeridos
en el ciclo celular. Estos mecanismos de control pueden ser activados por
diferentes señales fisiológicas que pueden actuar sobre diferentes complejos
CDK-ciclina .
<Puntos de control
y el proceso de tumorigénesis
La replicación y segregación del ADN, de los centriolos y
de los polos ecuatoriales están finamente regulados. Defectos en estos
mecanismos resultarán en formas de inestabilidad genómica como deles iones,
amplificaciones, translocaciones, no disyunción de los cromosomas y cambios en
la polaridad del genoma. Estas aberraciones se presentan durante la evolución
de las células normales hacia células con potencial tumorigénico.
Los puntos de control del ciclo celular tienen una
función importante en el mantenimiento de la fidelidad e integridad de la
replicación y reparación del genoma. La dinámica del ciclo celular está regulada
por estos puntos de control que actúan en la transcripción de los genes de CDK
y de las ciclinas, en las modificaciones postranscripcionales de estas
proteínas, o en la degradación de las mismas. Los procesos de regulación por
retroalimentación positiva y negativa también contribuyen a la progresión del
ciclo celular. Los controles negativos en dicha progresión están presentes
durante el desarrollo, diferenciación, senescencia y muerte celular, y pueden
tener una función importante en la prevención de la tumorigénesis. Se conocen
dos estadios donde operan los puntos de control en el ciclo celular: uno al
final de la fase G1 y la entrada a la fase S, y el otro, en la transición de la
fase G2 a la fase M. De manera general, en la mayoría de los casos, la
interrupción de la proliferación celular ocurre cuando la integridad del genoma
ha sido comprometida. Alteraciones en el proceso de interrupción del ciclo
celular permiten que células con genomas inestables evolucionen a células
cancerosas. Tales circunstancias podrían incluir: la senescencia celular, en
donde los telómeros (secuencias repetidas que están en los extremos de los
cromosomas) se pierden o llegan a ser cortos y se forman los cromosomas
dicéntricos inestables; la muerte celular por apoptosis, donde las nucleasas
que degradan al ADN están alteradas, y la naturaleza de la respuesta inmune del
hospedero, donde se requiere el rearreglo de genes de inmunoglobulinas o del
receptor de antígenos de linfocitos T, entre otros casos. No obstante, las
células tienen la capacidad de detener la progresión del ciclo celular en una
fase específica, cuando el daño es inducido por agentes extrínsecos que inhiben
la replicación del ADN. Los genes que codifican para proteínas que participan
en esta detención y que e stablecen la dependencia del ciclo celular son los
que constituyen los puntos de control del ciclo, regulando los procesos de
replicación, activación transcripcional, progresión del ciclo celular y
apoptosis.
Función de p53 en
el control del ciclo celular
La p53 es un transregulador transcripcional conocido como
un gen supresor de tumores. La proteína presenta tres dominios: el N-terminal,
que activa la transcripción; el central hidrofóbico, con regiones conservadas
que al mutar alteran la capacidad de unión al ADN y su actividad como factor
transcripcional, y el C-terminal, que participa en la oligomerización y unión
específica al ADN. Entre las funciones más importantes de p53 se encuentran su
capacidad para regular la transcripción de genes que participan en el control
del ciclo celular. Mutaciones de p53 pueden inducir cambios en el ciclo celular
y, por lo tanto, contribuir al desarr ollo de cáncer. En varios estudios, se ha
encontrado a p53 mutado asociado a diversas neoplasias (cuadro I).
La proteína p53 funciona como un regulador negativo del
ciclo celular, por lo que alteraciones en el gen que interfieren con su función
conducen a la pérdida de esta regulación, lo que produce una rápida
proliferación celular. La pérdida de la función de p53 está asociada con la
inmortalización y/o transformación in vitro in vivo. Se ha propuesto que p53
funciona como un punto de control para regular el paso de las células de un
estado de reposo a otro de proliferación. Esto se observa cuando las células se
exponen a agentes que dañan el ADN. Se sabe que la elevación de los niveles de
p53 induce a que las células se detengan al final de la fase G1 y se reparen los
daños en el ADN propiamente por la maquinaria de reparación de éste, antes de
continuar con s u replicación en la fase S. Las células con p53 mutado no
interrumpen el ciclo celular aun después de que el ADN ha sufrido daño.16 y al
desarrollo de neoplasias
Como ya se mencionó, se conocen dos estadios donde operan
los puntos de control en la progresión del ciclo celular: en relación con el
primero, al final de la fase G1 y la entrada a la fase S del ciclo celular, p
53 tiene una función central, ya que aumenta los niveles de los complejos
CDK-ciclina, que a su vez modulan la expresión de genes que participan en la
proliferación celular, específicamente en la interrupción del paso de la fase
G1 a la fase S, y esto permite la reparación del ADN dañado antes de que
continúe el ciclo celular. Además, hay evidencias que demuestran que p53
interactúa con los complejos CDK-ciclina. De esta manera, p53 puede reprimir la
expresión de genes que participan en los procesos de replicación y
transcripción del ADN, como es el caso del antígeno nuclear de proliferación
celular (PCNA), B-m y b, la ADN polimerasa a, C-fos, C-jun, MDM2; o bien,
activa genes reguladores negativos de la proliferación celular como Rb,
WAF1/CIP1/SD11, GADD45 y GADA, produciendo interrupción del ciclo celular o
muerte por apoptosis.9,20,21 En relación con el segundo estadio, donde operan
los puntos de control y que comprende la transición de la fase G2 a la fase M
del ciclo celular, se tiene poca información. Los puntos de control en fases
tempranas del ciclo están representados por los complejos CDK-ciclina. Por
ejemplo, las actividades de CDK2-E y CDK2-A son inhibidas por radiación
ionizante en una manera dependiente de p53, mediante la activación
transcripcional de p2122 (figura 2).
Existen evidencias que sugieren que alteraciones en p53 y
en los puntos de control producen inestabilidad genómica y sobrevivencia
inapropiada de células dañadas, que contribuyen a la evolución de células
normales a células malignas. Entre estos hallazgos se pueden mencionar: a) el
hecho de que p53 se encuentra mutado en muchos tipos de cánceres, lo que
sugiere que anormalidades en los puntos de control de la fase G1 a la fase S
son importantes en la tumorigénesis; b) las aneuploidias y las amplificaciones
de genes son comunes en células mutadas en p53, lo que sugiere que la pérdida
de la función de p53 está asociad a la inestabilidad genómica; c) los productos
de genes virales relacionados con cáncer (SV40, VPH y adenovirus) alteran la
función de varias proteínas celulares incluyendo a p53 y Rb, y pueden afectar
la función de los puntos de control que operan en el paso de la fase G1 a la
fase S del ciclo celular. El complejo entre la proteína transformante del virus
de papiloma humano E6 y p53 conduce a una rápida degradación de p53 mediante
proteólisis dependiente de ubiquitina, y por lo tanto, se pierde la
interrupción del ciclo celular inducido por p53;26 d) los pacientes con ataxia
telangiectasia tienen inestabilidad genómica, alta incidencia de linfomas
linfoblásticos27 y presentan alteraciones en genes que se requieren para la
inducción óptima de p53 en la fase S después de la irradiación, y e) en
adenocarcinomas de epitelio esofaríngeo se ha sugerido una función de los
mecanismos que controlan el paso de la fase G1 a la fase S dependiente de p53.
Además, la expresión anormal de las ciclinas D, E y A, en asociación con varias
CDK alteradas, en células deficientes de la función de p53, sugiere un
mecanismo adicional de la pérdida de los puntos de control durante la
transición de la fase G1 a la fase S en el proceso de tumorigénesis.
En algunos tejidos y bajo ciertas condiciones
fisiológicas, la inducción de p53 por daño al ADN causa muerte celular por
apoptosis, en lugar de interrupción del ciclo celular en la fase G1. En estas
instancias, la pérdida de la capacidad para que las células mueran por
apoptosis puede contribuir a la inestabilidad genómica y a la tumorigénesis y,
en consecuencia, a la pérdida del mecanismo de eliminación de células con daño
génico. Esto ocurre tempranamente en la progresión del cáncer y permite la
inestabilidad genómica con la sobrevivencia de células dañadas, o bien, ocurre
tardíamente en la tumo rigénesis y contribuye a la sobrevivencia de las células
en situaciones fisiológicas inapropiadas. La selección negativa en el tejido
tímico ocurre por apoptosis, y alteraciones en este mecanismo contribuyen al
desarrollo de linfomas linfoblásticos.
Por ejemplo, el oncogén Bcl-2, asociado a linfomas
granulocíticos, bloquea la apoptosis mediada por p53 después de la irradiación
de timocitos y otros tipos celulares (cuadro I). Además, el oncogén celular
c-myc y el gen de adenovirus E1A, pueden simultáneamente participar en la
proliferación celular y la apoptosis. Así, el proceso de apoptosis puede estar
regulado por genes que controlan la progresión del ciclo celular, lo que puede
resultar en el aumento de la inestabilidad genómica y de la sobrevivencia de
células transformadas.
Función de Rb en
el control del ciclo celular
El otro transregulador transcripcional del ciclo celular
es Rb, originalmente alterado en individuos con retinoblastoma. Rb es un gen
supresor de tumores que inhibe la proliferación celular. Esto se ha demostrad o
en células tumorales que carecen de este gen, y al momento de transfectarlas
con Rb, se observa una supresión del potencial tumorigénico.32 Además, se ha
observado que un exceso de Rb puede inhibir la proliferación celular aun en
células normales.33 No se ha reportado que Rb tenga una regulación
transcripcional, por lo que este gen se expresa constitutivamente en células en
división o en estado de reposo. Por lo tanto, la regulación de Rb ocurre a
nivel postranscripcional, por fosforilación de la proteína (pRb). Aunque
directamente la función de pRb no se asocia con la interrupción del ciclo
celular, existen evidencias de que la forma no fosforilada de pRb es
responsable de la interrupción de la proliferación celular. Además, la
fosforilación de esta proteína es crucial para su unión con otras proteínas
involucradas en la activación transcripcional y en la regulación del ciclo
celular.
Para determinar la función de pRb, se ha estudiado su
estado de fosforilación durante el ciclo celular. En las fases G0 y G1
tempranas del ciclo, pRb se encuentra hipofosforilada (forma activa). La
fosforilación de pRb ocurre entre las fases G1 y S, y aumenta en las fases G2 y
M (forma inactiva). Cuando la célula termina la mitosis pRb se defosforila. La
actividad de pRb está principalmente asociada a inhibición de la proliferación
celular por contacto célula-célula, por falta de estímulos proliferativos o por
la presencia de estímulos antiproliferativos como TGF-b1 o TNF-a. En células
normales en reposo, pRb se encuentra hipofosforilada y asociada al factor transcripcional
E2F. Los diferentes estados de fosforilación de pRb durante el ciclo celular
sugieren que pRb es un sustrato de los complejos CDK-ciclina. La expresión,
fosforilación y formación de los complejos CDK-ciclina durante la fase G1
tardía, están asociados a la inactivación de pRb. Por ejemplo: CDK2 se une con
pRb y la fosforila en residuos de serinas o treoninasin vivo. CDK4-D se une y
fosforila pRb in vitro. Las ciclinas E y D se acumulan en la fase G1 tardía, y
su expresión corresponde con el momento en el que ocurre la fosforilación de
pRb, mientras que la expresión de la ciclina A ocurre en la fase S temprana y
en la fase G2, cuando pRb se encuentra hiperfosforilada (figura 2). En general,
las ciclinas D2 y D3 son más estables para formar complejos específicos con pRb
que las ciclinas A y E.40 Además, en células con daño genético, la transfección
con pRb induce interrupción de la proliferación celular en la fase G1; sin
embargo, al realizar una cotransfección con las ciclinas A o E, se libera el arresto
celular inducido por pRb y se produce hiperfosforilación de pRb.
En tumores humanos donde pRb está mutada, ésta no se
fosforila y pierde la capacidad de suprimir la proliferación celular. Esta
mutación impide la unión de pRb a factores de proliferación celular e incluso
la unión con oncoproteínas virales, por
lo que pRb puede actuar como un precursor de tumores al no inducir la
interrupción del ciclo celular. No obstante, existen evidencias sobre la
interrupción del ciclo celular por pRb al modular la actividad de varios
factores de transcripción. Normalmente, el factor de elongación de la
transcripción E2F forma un complejo con pRb no fosforilada e impide la
transcripción de genes requeridos en la fase S. Estos complejos pRb-E2F pueden
ser disociados por varias oncoproteínas virales como E1A, e incluso ésta
disocia los complejos de ciclina A, CDK2 y p107 unidos a pRb, que se forman
durante la fase S. Por lo tanto, la formación de complejos entre pRb y E2F
puede ser interferida por proteínas virales como E1A de adenovirus, E7 del
virus de papiloma humano y antígeno T de SV40, que son capaces de un irse a pRb
no fosforilada (figura 2). El mecanismo de regulación por parte del complejo
pRb-E2F ya ha sido bien caracterizado.
Finalmente, pRb no sólo actúa como gen supresor de
tumores, sino también como activador de la transcripción de genes que suprimen
la proliferación celular. Por ejemplo, la transfección con Rb activa la
transcripción de los genes TGF-b1 y TGF-b2 en queratinocitos, factor que
interrumpe la progresión del ciclo celular en la fase G1, mientras que pRb
permanece defosforilada.48 Además, se conoce que pRb puede participar en el
proceso de apoptosis mediada por p53.
Activación celular
y puntos de control del ciclo celular
La activación celular es un programa finamente regulado
en donde participan una gran cantidad de elementos los cuales son regulados por
varios mecanismos de control. En la mayoría de los casos, la activación celular
comienza con la interacción del ligando con el receptor correspondiente (por
ejemplo antígeno-receptor de linfocitos T, EGF-EGFR, IL-2-IL2R, etc.). La
transducción de señal se continúa en el interior celular con la activación de
proteínas tirosinas cinasas (Fyn, Lyn, Lck, ZAP-70, etc.) que se encuentran
acopladas a los dominios intracelulares de los receptores. La estimulación del
receptor también puede involucrar la activación de la fosfolipasa C (PLC) que
hidroliza al fosfatidil inositol bifosfato (PIP2) y genera inositol trifosfato
(IP3) que mobiliza iones Ca++, y diacilglicerol (DAG) que a su vez activa a la
proteína cinasa C (PKC). La inducción de tirosinas cinasas y PKC activan a los
miembros de la familia de Ras (Ash, Grb2, Ras, Sos, etc.), los cuales trasmiten
la señal al núcleo a través de la activación de proteínas cinasas activadas por
mitógeno (MAPKK, MAPK, JNKK, JNK) y se lleva a cabo la activación
transcripcional de muchos genes a través del reconocimiento de elementos de
respuesta específicos, como por ejemplo AP-1 y SRE. Otra vía de transducción es
la de adenilato ciclasa (AC), la cual se encuentra asociada a receptores que al
ser activados producen AMPc. El AMPc activa la proteína cinasa A (PKA) para
generar la proteína de unión al elemento de respuesta a AMPc (CREB). En las si
tuaciones donde existe daño genético se induce a p21 a través de la forma
activa de p53. Entre las funciones de p21 se encuentra la disociación de los
complejos CDK-ciclina y en consecuencia s e interrumpe el ciclo celular. La
formación de los complejos CDK2-E activos disocia a los complejos pRB-E2F
liberando a E2F, lo cual influye en la activación transcipcional y en la
progresión del ciclo celular (figura 2).
Como puede apreciarse, existe una estrecha asociación
entre el proceso de activación celular y los puntos de control del ciclo
celular. Los puntos de control en la transición de la fase G1 a la fase S y de
la fase G 2 a la fase M son importantes en la protección de las células de
fuentes exógenas de daño al ADN. Sin embargo, este daño puede ser causado por procesos
celulares intrínsecos como el rearreglo de genes durante el d esarrollo, la
senescencia celular y la muerte celular por apoptosis. Cuando el daño es
generado por procesos intrínsecos, los puntos de control sobre la proliferación
celular son importantes en la prevención de la evolución de las células
normales a cancerosas. Se ha informado una asociación entre el aumento de edad,
la incidencia de cáncer, el aumento de daño al ADN debido a una exposición
acumulada de agentes que lo dañan y la disminución de la capacidad de
reparación del ADN. Por otra parte, los estudios de senescencia celular han
revelado una importante fuente de daño celular intrínseco. Los fibroblastos
humanos normales no expresan telomerasa; por lo tanto, los telómeros decrecen
con la proliferación celular. Se ha sugerido que la senescencia celular está
asociada a la pérdida de secuencias teloméricas y que los cromosomas con telómeros cortos
activa n puntos de control que inhiben la proliferación celular. Las células
senescentes tienen un aumento de aberraciones cromosómicas, con asociaciones
telómero-telómero, por lo que el programa senescente normal puede gener ar
inestabilidad genómica. Un gen que es necesario para interrumpir la
proliferación en células senescentes es p21 (WAFI/CIPI/SDII, cuyo producto se
une a los complejos CDK-ciclina e inhibe sus funciones. La pérdida de p21 en bilidad
genómica (figura 1).
Durante el rearreglo de genes de inmunoglobulinas y del
receptor de linfocitos T se generan rupturas del ADN y hay proteínas que
inhiben la progresión del ciclo celular durante estos procesos. Por lo tanto,
los genes que codifican estas proteínas son blancos potenciales para mutaciones
que podrían generar inestabilidad genómica. Estas mutaciones son importantes en
la etiología de linfomas y leucemias. El escape de la interrupción de la
proliferación celular, por mutación en genes que controlan la proliferación
durante la apoptosis, induce la proliferación de células con inestabilidad
genómica, que estaban comprometidas a morir.
Por otra parte, el funcionamiento inapropiado del uso
mitótico induce interrupción de la progresión del ciclo celular. Además, hay
inhibición de un nuevo ciclo si la mitosis no fue completada en el ciclo previo
debido a la inhibición del ensamblaje de microtúbulos. Se ha informado que las
células cancerosas tienen un aumento en la resistencia a agentes
antimicrotúbulos en relación con las células normales. La regulación de los
centriolos ha sido menos estudiada; sin embargo, defectos en su duplicación inducen
detención de la mitosis por medio de un punto de control. Por ejemplo, la
expresión del antígeno T del virus SV40 e n tejido pancreático murino produce
anormalidades en el número y segregación de centriolos, y esto a su vez genera
inestabilidad genómica. El producto del proto-oncogén c-mos, un regulador de la
metafase meiótica, produce poliploidia cuando se expresa anormalmente en
células mitóticas, así como durante la tumorigénesis .
Respecto a la transición de la fase G2 a la fase M, ésta
es inhibida por el daño y replicación alterada del ADN. Los puntos de control
evitan la segregación de cromosomas alterados. Se han identificado pocos genes
que controlan la transición de la fase G2 a la fase M; sin embargo, los
defectos en la regulación de los puntos de control que actúan en esta etapa,
pueden ser importantes en la tumorigénesis. Por ejemplo, se ha de mostrado que
la sobrexpresión de Ras normal o mutado promueve la formación de células
multinucleadas y desórdenes mitóticos, lo que sugiere que Ras puede participar
en la desregulación de la transición de la fase G2 a la fase M del ciclo
celular. Las células de individuos con predisposición a cáncer familiar
muestran mayor frecuencia de rupturas cromosómicas después de la irradiación.
Las células de pacientes con ataxia telangiectasia se detienen en la fase G1
después de la irradiación. Líneas celulares derivadas de cánceres humanos
interrumpen su progresión en la fase G2 después del daño al A DN. Además, la
expresión alterada de elementos, que participan en la transición de la fase G2
a la fase M, como las ciclinas A, B y de CDK2, se presenta en los mismos
cánceres.
Control del ciclo
celular y terapia contra el cáncer
En términos generales, en las estrategias instrumentadas
contra el desarrollo del cáncer, el efecto que produce la mayoría de los
agentes antineoplásicos es daño al ADN, al aparato mitótico, a las
topoisomerasas, o inhiben la síntesis o incorporación de precursores del ADN.
El éxito de estos agentes en la muerte selectiva de las células cancerosas
varía principalmente en función del tipo de cáncer. Algunos cánceres son
sensibles a estos agentes y son curables (leucemia linfoblástica aguda y
cánceres de células germinales), mientras que otros son relativamente
resistentes y no son curables (carcinoma de colon). Esta variabilidad de
respuestas refleja la especificidad celular ante los agentes anticancerígenos.
En consecuencia, los puntos de control del ciclo celular representan una buena
opción para la aplicación de los agentes quimioterapéuticos.
En este contexto, varias propiedades importantes de los
puntos de control del ciclo celular merecen cierta consideración.
1. Los puntos de control son sistemas de transducción de
señal. La existencia de varios componentes para cada uno de los puntos de
control sugiere la presencia de cascadas de transmisión, y cada una de éstas
representa múltiples blancos para la intervención terapéutica.
2. La mayoría de los genes de los puntos de control son esenciales
en células normales, ya que ellos codifican para componentes de la maquinaria
del ciclo celular que emiten o suprimen señales, o porque participan en más de
una función celular. Para las vías no esenciales, los agentes terapéuticos que
están dirigidos hacia los puntos de control sólo serán detectados por su sinergismo
con otros agentes que dañen a la célula.
3. Los puntos de control aseguran la fidelidad e
integridad de la replicación y segregación genómica. La reparación del daño
espontáneo del ADN requiere del correcto funcionamiento de los puntos de
control para que las células mantengan esta fidelidad e integridad en la
replicación del genoma. Por lo tanto, la restauración de los puntos de control
que están alterados podría retardar la evolución de la célula normal a célula
cancerosa, de igual manera que en ausencia de fuentes exógenas que dañan al
ADN.
4. Los sistemas de transducción de señal exhiben
adaptación. Si los puntos de control están dañados, las células prosiguen el
ciclo celular aun cuando la alteración no haya sido reparada. Las alteraciones
génicas que aumentan la habilidad de las células para adaptarse podrían
acelerar la evolución del proceso neoplásico. Además, la inhibición de los
componentes involucrados en la adaptación sugiere blancos terapéuticos para
reparar los puntos de control alterados, como un mecanismo que retardaría la
evolución de las células normales a células precancerosas.
5. La activación de los puntos de control induce una
variedad de respuestas celulares. Por ejemplo, la expresión anormal de p53 en
células que entran a la fase S con el ADN dañado es más nociva, por el hecho de
que estas células no sufren muerte por apoptosis. La función de los puntos de
control en la apoptosis está influida por el tipo celular y por la naturaleza
de las señales de proliferación, o bien, por el daño al cual las células
responden. La restauración de los puntos de control podría inducir la respuesta
de muerte celular por apoptosis de las células cancerosas y aumentar la
sensibilidad a los agentes que daña n el ADN. De hecho, al aplicar terapia
génica con p53 normal a células de cáncer cervicouterino, se induce el proceso
de apoptosis. Por lo tanto, los componentes de la respuesta apoptótica podrían
ser usados como blancos terapéuticos, si la apoptosis pudiera ser activada en
ausencia del daño al ADN. Esto podría ser posible para probar la especificidad
de cierto tipo de células cancerosas, ya que no todas las células responden a
las mismas señales apoptóticas.
El conocimiento de nuevas drogas que inhiben o activan
puntos de control permite la designación de estrategias terapéuticas
eficientes. Para los cánceres localizados, la inhibición de un punto de control
no tiene efecto en las células que simultáneamente no se expusieron a agentes
que dañan el ADN, por lo que la radiación local de los cánceres o la acción de
agentes citotóxicos facilitará el uso de tales compuestos. Las células de
individuos con ataxia telangiectasia tienen alteraciones en los puntos de control
que regulan el ciclo celular en la transición de la fase G1 a la fase S, y de
la fase G2 a la fase M después de la radiación local, y son sensibles a efectos
citotóxicos de radiación. Por lo menos, uno de los genes responsables de ataxia
telangiectasia ha sido localizado en el cromosoma 11q, y éste podría ser un
blanco terapéutico idóneo.
Una característica que puede distinguir a los cánceres
que son curados con quimioterapia es la capacidad de sanar por un rápido
proceso de apoptosis, en respuesta a agentes citotóxicos. Algunos de los genes
que regulan la progresión del ciclo celular de la fase G1 a la fase S están
involucrados en el control de la apoptosis. Además, la muerte celular por
apoptosis ocurre como un balance del control positivo y negativo de las señales
de proliferación, lo cual sugiere asociaciones entre genes que controlan el
proceso de apoptosis y el ciclo celular. Estos genes son posibles blancos para
la manipulación de la sobrevivencia celular después de la exposición a agentes
citotóxicos. El producto del gen Bcl-2 protege a las células de la muerte por
apoptosis mediada por Bax. La caracterización de otras moléculas similares a
Bcl-2 o que interactúan con Bcl-2, tales como B cl-xl y Mcl-1, sugieren blancos
atractivos para el implemento de terapias anticancerígenas. Por otra parte,
mientras que los fibroblastos usan a p53 para interrumpir el ciclo celular
después del daño del ADN, los timocitos mu eren por apoptosis mediada por p53.
La pérdida de la función de p53 en las células que inician el proceso de
apoptosis produce resistencia al tratamiento citotóxico. Así, una terapéutica
global sería inducir el proceso de apoptosis, en lugar de interrumpir el ciclo
celular en las células cancerosas que tienen p53 mutado.
Los controles moleculares de la progresión del ciclo
celular pueden proveer nuevos blancos para agentes citotóxicos. Por ejemplo,
las células cancerosas mutadas en Rb tienen un aumento en los niveles de E2F-1,
lo cual teóricamente resultaría en el aumento de la transcripción de ciertos
genes como: dihidrofolato reductasa (DHFR), timidilato sintetasa (TS),
ribonucleótido reductasa (RR) y timidina kinasa (TK). El aumento de la expresión
de DHFR induce resistencia a metotrexate. De manera similar, el 5-fluorouracilo
u otros inhibidores de TS pueden ser efectivos, no sólo porque funcionan
regulando negativamente al DHFR, sino porque actúan uniéndose a la TS e inhiben
su actividad. Tal efecto podría ser usado en el osteosarcoma, en el cual Rb
está mutado, y el metotrexate es un agente antineoplásico comúnmente usado.
Además, el metabolismo de purinas se ha estudiado a mpliamente y representa
otra alternativa para el desarrollo de agentes antineoplásicos.
Nuevos productos génicos o productos de genes
sobrexpresados en cánceres específicos proveen otros blancos potenciales para
la terapia contra el cáncer. Por ejemplo, la proteína quimérica timidina cinasa
bcr-abl y el factor transcripcional AML-1, no están presentes en los tejidos
normales, pero se generan de translocaciones cromosomales en la leucemia
mielogénica crónica y en la leucemia mieloide aguda, respectivamente. Estas
proteínas contribuyen a la alteración del ciclo celular y el proceso de
apoptosis en las células malignas; por lo tanto, disparan la expresión de genes
específicos, afectando la mayoría de los puntos de control. La reciente
demostración de la expresión de telomerasa en células cancerosas pero no en
células normales, así como la potencial dependencia de las células cancerosas
de la actividad de la telomerasa para la viabilidad, supone a la telomerasa
como otro blanco atractivo para la terapia específica anticancerígena. La
pérdida de la telomerasa probablemente activa los puntos de control en la
transición de la fase G2 a la fase M, induce la interrupción del ciclo celular,
y probablemente, el proceso de apoptosis.
Control del ciclo
celular y prevención del cáncer
Si la inestabilidad genómica es una de las condiciones
para el desarrollo del cáncer, la reducción de esta inestabilidad ayudaría a
prevenirlo. Por ejemplo: p53 es la proteína de control del ciclo celular mejor
caracterizada, relacionada con la inestabilidad genética. Mutaciones en p53
parecen ocurrir en etapas tempranas de ciertos cánceres y pueden estar
presentes en displasias, pero no en lesiones malignas del epitelio bronquial.
La exposición de tales células a agentes que dañan el ADN puede explicar el
aumento en la frecuencia de aberraciones génicas. La prevención del daño al ADN
evitaría tales aberraciones. No todo el daño al ADN es causado por agentes
exógenos; también se presenta intracelularmente daño oxidativo que conduce a la
célula de normal a maligna. Por lo tanto, la prevención del daño oxidativo
posiblemente sería efectiva en el desarrollo del cáncer para retardar o evitar
la transformación maligna. Los agentes ambientales que alteran los puntos de
control del pase de la fase G1 a la fase S, aumentan la inestabilidad genómica
de igual manera que el daño oxidativo. La identificación y/o eliminación de los
agentes ambientales que actúan inhibiendo los puntos de control o reparación
del ADN, pueden ser una estrategia de prevención efectiva contra el cáncer. Los
cánceres de cabeza, cuello, orales, esofaríngeos y de la piel presentan
mutaciones en p53, y todos los tejidos tienen probablemente una exposición
significativa a agentes que dañan el ADN. Alternativamente, si las células
cancerosas premalignas retienen a p53 normal, la inducción del proceso de
apoptosis podría retardar el porcentaje de progresión a un estado maligno. En
células donde están alterados los puntos de control, la restauración de la
función de éstos es otro mecanismo potencial para abolir la progresión del
cáncer, pero es una de las tareas más difíciles de realizar. Sin embargo, las
estrategias para la manipulación de la función de p53 incluyen técnicas de
transfección de genes (terapia génica) que inducen la expresión de p53 para
restaurar su función normal. Estos resultados sugieren que la transfección con
p53 normal a células con cáncer cervical representa una potencial estrategia
para la terapia de este tipo de cáncer. La inactivación de la proteína MDM2 es
otro blanco potencial para restaurar la función de p53.
Bibliografías:
·a) Patología estructural y
funcional. Octava edición. Editorial El sevier Saunders. Autores: Kumar, Abbas,
Fausto, Aster. Paginas consultadas 279-282.
· b)Brandan, Nora y Juaristi,
Julián, Profesores Titulares. Cátedra de
Bioquímica. Facultad de Medicina. UNNE.
Consultado el: Disponible en ht Judith Meza Junco, Aldo Montaño Loza;
Álvaro Aguayo González. Revista de Investigación clínica. Rev. Invest. Clin.
2014(25 de marzo de 2014);v.58 n.1. Disponible en: http://www.scielo.org.mx/scielo.php?pid=S0034-83762006000100008&script=sci_arttext